


SARS-CoV-2-like Spike Protein Sequences with Affinity to hACE2 Were Present in the Public Databases in 2005. Plus, a Hint on SARS-CoV-2 Laboratory Origin
Lau et al. published three sequences with an interesting legacy that involves genetic manipulation by Ralph Baric published in 2008. The implications are profound; Why is everyone ignoring it?

In 2020, I published an analysis of all of the available SARS-CoV-2 and SARS sequences at the time in search of a specific relationship. If any SARS sequences published prior to 2020 were similar to SARS-CoV-2 with respect to functional motifs, then the question of origins of SARS-CoV-2 would have to address how a SARS-CoV-2 like spike protein made its way back through time to when the older SARS sequences were being published.
A motif describes a short amino acid arrangement that is shared by protein family members. They are useful in combination with information with amino acid sequence (aka protein) sequence databases to assign putative functions to unknown proteins.
Then there's a study from 2008 where a SARS-like RNA sequence was derived so it would encodes a consensus genomic sequence derived from 4 viral sequences deposited into the databases was made to infect murine (mouse) cell lines. This study was conducted by UNC’s Ralph Baric and colleagues:
Becker MM, Graham RL, Donaldson EF, Rockx B, Sims AC, Sheahan T, Pickles RJ, Corti D, Johnston RE, Baric RS, Denison MR. Synthetic recombinant bat SARS-like coronavirus is infectious in cultured cells and in mice. Proc Natl Acad Sci U S A. 2008 Dec 16;105(50):19944-9. doi: 10.1073/pnas.0808116105. Epub 2008 Nov 26. PMID: 19036930; PMCID: PMC2588415.
The work described here was the type of direct laboratory viral genome manipulation and editing that was being denied to have occurred in WIV in spite of evidence otherwise. From their Results section:
Consensus Bat-SCoV Sequence Design and Construction.
“When this study was initiated, 4 Bat-SCoVs had been identified (HKU3–1, HKU3–2, HKU3–3, and RP3) as the virus reservoir populations from which SARS-CoV emerged (10–12). Because none had been recovered in culture, the infectivity of the reported viral genomic RNA sequences was hypothetical, having been derived from RT-PCR sequencing of bat fecal or rectal swab samples. Sequence databases have error frequencies from 1/500 to 1/10,000, making viable genome reconstruction problematic with increasing size (23). Therefore, we used the 4 reported Bat-SCoV sequences to establish a putative consensus Bat-SCoV sequence (GenBank accession no. FJ211859) and designed cDNA fragments with junctions precisely aligned to the existing SARS-CoV reverse genetics system [Fig. 1A; supporting information (SI) Fig. S1] (24). The defined and functional SARS-CoV 5′ UTR and transcriptional regulatory sequences were used because the 5′ UTRs of the Bat-SCoVs were incomplete. The genomic cDNA fragments were commercially synthesized, inserted into vectors, assembled into a full-length cDNA, and transcribed in vitro to yield genomic RNA. Initial attempts to recover and passage infectious Bat-SCoV failed. Electroporated cells contained high levels of genome and leader-containing subgenomic transcripts on day 2, but not day 5 postelectroporation (p.e.) (Fig. 1C), indicating that the synthetic consensus Bat-SCoV genome expressed a functional replicase. We did recover infectious virus consisting of SARS-CoV genome fragments A–E and Bat fragment F (Fig. 1 B and D). The resulting virus, Bat-F, encoded a chimeric Spike. Thus, the amino-terminal two-thirds of SARS-CoV Spike, including the RBD, and the fusion core contained within the carboxyl-terminal third of Bat-SCoV Spike can successfully drive productive infection. Also, because Bat-F contained Bat-SCoV accessory and structural genes 3′ to the Spike gene, these downstream ORFs are clearly interchangeable.

Schematic representation of SARS-CoV and Bat-SCoV variants. (A) Schematic representation of SARS-CoV and Bat-SCoV (GenBank accession no. FJ211859) genomes and reverse genetics system. (Top) Arrowheads indicate nsp processing sites within the ORF1ab polyprotein (open arrowheads, papain-like proteinase mediated; filled arrowheads, nsp5 [3C-like proteinase] mediated). Immediately below are the fragments used in the reverse genetics system, labeled A through F. The fragments synthesized to generate Bat-SCoV exactly recapitulate the fragment junctions of SARS-CoV with the exception that the Bat-SCoV has 2 fragments, Bat-E1 and Bat-E2, which correspond to the SARS-E fragment. (B) Schematic representation showing organization of the SARS-CoV and Bat-SCoV Spike proteins. The engineered Spike proteins are pictured below with the virus name to the left. Bat-SRBD includes all of the Bat-SCoV Spike sequence except that the Bat-SCoV RBD (Bat-SCoV amino acid 323–505) is replaced with the SARS-CoV RBD (amino acid 319–518) (GenBank accession no. FJ211860). Bat-SRBD-MA includes the MA15 Spike RBD change at SARS-CoV aa Y436H. Bat-SRBM includes the minimal 13 SARS-CoV residues critical for ACE2 contact, resulting in a chimeric RBD of Bat-SCoV amino acid 323I-429T and SARS-CoV amino acid 426R-518D. Bat-Hinge is Bat-SRBM sequence, with Bat-SCoV amino acid 392L-397E replaced with SARS-CoV amino acid 388V-393D. Bat-F includes nt 1–24057 of SARS-CoV (to Spike amino acid 855), with the remaining 3′ sequence from Bat-SCoV. To the right of the schematic representations, observation of transcript activity and approximate stock titers at passage 1 (P1) are indicated. ND indicates no infectious virus detected by plaque assay. (C and D) Presence of genomic and subgenomic transcripts after electroporation of in vitro transcribed viral RNA. Band corresponding to mRNA1 indicates the presence of genomic RNA, either electroporated genomic RNA or progeny genomic RNA, and the presence of a band corresponding to mRNA9 indicates the presence of leader-containing subgenomic RNA, consistent with mRNA transcription.
A careful read shows that Baric and colleagues were directly interested in characterizing the hACE2 (human ACE2) receptor binding capacity of SARS viruses:
“The ectodomain of Spike can be exchanged among CoVs, altering host-range specificity (25, 26). To test whether the RBDs of Bat-SCoV and SARS-CoV were interchangeable, we replaced the Bat-SCoV RBD (amino acid 323–505) with the SARS-CoV RBD (amino acid 319–518) (27, 28) (GenBank accession no. FJ211860), simulating a theoretical recombination event that might occur during mixed infection in vivo (Fig. 1B). After electroporation, Bat-SRBD genome RNA and leader-containing subgenomic mRNA transcripts were detected (Fig. 1C), and progeny virions were detected by plaque assay. After 2 additional passages, the population genome sequence was identical to the Bat-SRBD molecular clone. However, 4 nucleotides exhibited dual peaks on the sequencing electropherograms, suggesting quasispecies variation at these positions (Table S1). Recovery and passage of Bat-SRBD demonstrated the functional interchangeability of human and animal SARS-CoV-like RBDs.
The crystal structure of SARS-CoV RBD complexed with its receptor, hACE2 (29), implicated 13 residues within the carboxyl terminus of the RBD (amino acid 426R-518D) in ACE2 engagement. Homology modeling indicated that this receptor-binding motif (RBM) may be sufficient to allow ACE2 engagement, and further predicts that inclusion of 6 residues amino-terminal to the RBM (amino acid 388V-393D) may enhance ACE2 engagement by functioning as a distal “hinge.” To test this possibility, chimeric Bat-SCoV genomes were constructed containing either the SARS-CoV RBM (Bat-SRBM) or the RBM plus the distal hinge residues (Bat-Hinge) (Fig. 1B). Electroporation yielded genome and subgenomic leader-containing transcripts at day 2, but not 5, p.e. (Fig. 1 C and D), and progeny virions could not be successfully passaged in culture.
I found this study by comparing the motif patterns of hundreds of “SARS” spike protein sequences to the motif pattern I had found for SARS-CoV-2 spike protein. Out of the hundreds I analyzed, two a SARS-CoV-2-like motif pattern. The database entries for HKU-3-1, HKU-3-2 and HKU-3-3 (data housed at NCBI) led me to the Baric et al. study.
HKU-3-1 and HKU-3-3 are not SARS. Baric et al. Did Not Know This. No One Did.
When I read the UNC publication tied to the SARS-CoV-2-like sequence, I was stunned. The study was based on four downloaded amino acid sequences. One of the 4 sequences used, Rp3, (the odd man out) had a different 3' protein motif pattern compared to the other three. That sequence has a SARS-like spike protein when the functional domain motifs are considered.
Baric and his colleagues clearly did not appreciate how different Rp3 was from HKU3-1, HKU3-2, and HKU3-3 in 2008 in terms of the spike protein receptor biology, otherwise they would not have calculated a consensus sequence.
The other three sequences, the HKU-3’s share an identical 3' functional motif pattern with the SARS-CoV-2 sequence. The sequences was deposited in Genbank in 2005; I found and reported this in 2020.
There are couple of things that need to be explained:
(1) The HKU sequences deposited by the Chinese researchers in 2005, from samples from the anus of a bats in Hong Kong, were downloaded and analyzed by Ralph Baric or someone working with him for the 2008 publication. It has a spike protein that is by far more similar to SARS-CoV-2 than to any SARS with respect to functional domain architecture.
(2) The sequences of interest do not contain the PPAR furin cleavage site motif that facilitates hACE2 receptor entry, but they have hACE2 binding affinity. We know this because
(3) The sequence constructed from the consensus of the wild-caught “SARS”-like viruses did have some affinity for hACE2. This was 2005.
(4) Baric and colleagues describe how the consensus lab-produced SARS mRNA was manipulated in the lab and made able to be better able to infect cell lines (clear gain-of-function research).
In 2020, I addressed the fact that SARS-CoV-2 like sequences (HKU3’s) were present in a database in 2005. Thus, the ideas that SARS-CoV-2 was made in the lab from SARS-virus is incorrect. If a sequence was manipulated or pushed through serial passage, the precursor must have been a ‘SARS-CoV-2 like’ sequence with the functional motif patterns I reported in 2020.
The Pathogenicity Motif Pattern
In my report from 2020, I reported that the HKU-3 SARS-CoV-2-like spike protein sequences contained a unique shortened N-terminal spike domain and a C-terminal Gp41 (retroviral envelope) motif. Neither of these features is found in SARS spike protein sequences. Further, in contrast to SARS, there is neither a She-3 or KxDL motif in the spike 2 segment. SARS has a few additional motifs of unknown function.
The results are easily reproduced using the the Motif Search web application (available at https://www.genome.jp/tools/motif/) using the FASTA-formatted spike protein amino acid sequences (or any amino acid sequences.
Any protein, or any part of a protein sequence can be analyzed using the Motif Search web application. It’s so simple everyone reading this article can execute the analysis. But that does not mean the information is simple. The rich information in the match of the motif elements is not expected - at all - given the official narrative of the evolution of beta-coronaviruses published to date.
Here’s the Protein Database entry for the HKU-3-3 sequence. It is SARS-CoV-2 like, not SARS-like, and the literature that refers to it as SARS-like is misleading.
>AAZ41340.1 spike glycoprotein [Bat SARS coronavirus HKU3-3]
MKILIFAFLANLAKAQEGCGIISRKPQPKMAQVSSSRRGVYYNDDIFRSDVLHLTQDYFLPFDSNLTQYF
SLNVDSDRYTYFDNPILDFGDGVYFAATEKSNVIRGWIFGSSFDNTTQSAVIVNNSTHIIIRVCNFNLCK
EPMYTVSRGTQQNAWVYQSAFNCTYDRVEKSFQLDTTPKTGNFKDLREYVFKNRDGFLSVYQTYTAVNLP
RGLPTGFSVLKPILKLPFGINITSYRVVMAMFSQTTSNFLPESAAYYVGNLKYSTFMLRFNENGTITDAV
DCSQNPLAELKCTIKNFNVDKGIYQTSNFRVSPTQEVIRFPNITNRCPFDKVFNATRFPNVYAWERTKIS
DCVADYTVLYNSTSFSTFKCYGVSPSKLIDLCFTSVYADTFLIRSSEVRQVAPGETGVIADYNYKLPDDF
TGCVIAWNTAKHDTGNYYYRSHRKTKLKPFERDLSSDDGNGVYTLSTYDFNPNVPVAYQATRVVVLSFEL
LNAPATVCGPKLSTELVKNQCVNFNFNGLKGTGVLTSSSKRFQSFQQFGRDTSDFTDSVRDPQTLEILDI
SPCSFGGVSVITPGTNASSEVAVLYQDVNCTDVPTAIRADQLTPAWRVYSTGVNVFQTQAGCLIGAEHVN
ASYECDIPIGAGICASYHTASVLRSTGQKSIVAYTMSLGAENSIAYANNSIAIPTNFSISVTTEVMPVSM
AKTAVDCTMYICGDSLECSNLLLQYGSFCTQLNRALTGIAIEQDKNTQEVFAQVKQMYKTPAIKDFGGFN
FSQILPDPSKPTKRSFIEDLLFNKVTLADAGFMKQYGDCLGDVSARDLICAQKFNGLTVLPPLLTDEMVA
AYTAALVSGTATAGWTFGAGAALQIPFAMQMAYRFNGIGVTQNVLYENQKLIANQFNSAIGKIQESLSST
ASALGKLQDVVNQNAQALNTLVKQLSSNFGAISSVLNDILSRLDKVEAEVQIDRLITGRLQSLQTYVTQQ
LIRAAEIRASANLAATKMSECVLGQSKRVDFCGKGYHLMSFPQSAPHGVVFLHVTYVPSQEKNFTTAPAI
CHEGKAYFPREGVFVSNGTSWFITQRNFYSPQLITTDNTFVSGNCDVVIGIINNTVYDPLQPELDSFKEE
LDKYFKNHTSPDVDLGDISGINASVVNIQKEIDRLNEVAKNLNESLIDLQELGKYEQYIKWPWYVWLGFI
AGLIAIVMVTILLCCMTSCCSCLKGACSCGSCCKFDEDDSEPVLKGVKLHYT

Every scientist interested in the origins of the SARS-CoV-2 virus to focus on HKU-3-3 as a putative ancestor, or relative of an ancestor, of the SARS-CoV-2 virus. The famous RatG13 sequence, alleged to be the “backbone” virus that was manipulated to become the SARS-CoV-2 virus, also has a SARS-CoV-2 like motif pattern.
>QHR63300.2 spike glycoprotein [Bat coronavirus RaTG13]
MFVFLVLLPLVSSQCVNLTTRTQLPPAYTNSSTRGVYYPDKVFRSSVLHLTQDLFLPFFSNVTWFHAIHV
SGTNGIKRFDNPVLPFNDGVYFASTEKSNIIRGWIFGTTLDSKTQSLLIVNNATNVVIKVCEFQFCNDPF
LGVYYHKNNKSWMESEFRVYSSANNCTFEYVSQPFLMDLEGKQGNFKNLREFVFKNIDGYFKIYSKHTPI
NLVRDLPPGFSALEPLVDLPIGINITRFQTLLALHRSYLTPGDSSSGWTAGAAAYYVGYLQPRTFLLKYN
ENGTITDAVDCALDPLSETKCTLKSFTVEKGIYQTSNFRVQPTDSIVRFPNITNLCPFGEVFNATTFASV
YAWNRKRISNCVADYSVLYNSTSFSTFKCYGVSPTKLNDLCFTNVYADSFVITGDEVRQIAPGQTGKIAD
YNYKLPDDFTGCVIAWNSKHIDAKEGGNFNYLYRLFRKANLKPFERDISTEIYQAGSKPCNGQTGLNCYY
PLYRYGFYPTDGVGHQPYRVVVLSFELLNAPATVCGPKKSTNLVKNKCVNFNFNGLTGTGVLTESNKKFL
PFQQFGRDIADTTDAVRDPQTLEILDITPCSFGGVSVITPGTNASNQVAVLYQDVNCTEVPVAIHADQLT
PTWRVYSTGSNVFQTRAGCLIGAEHVNNSYECDIPIGAGICASYQTQTNSRSVASQSIIAYTMSLGAENS
VAYSNNSIAIPTNFTISVTTEILPVSMTKTSVDCTMYICGDSTECSNLLLQYGSFCTQLNRALTGIAVEQ
DKNTQEVFAQVKQIYKTPPIKDFGGFNFSQILPDPSKPSKRSFIEDLLFNKVTLADAGFIKQYGDCLGDI
AARDLICAQKFNGLTVLPPLLTDEMIAQYTSALLAGTITSGWTFGAGAALQIPFAMQMAYRFNGIGVTQN
VLYENQKLIANQFNSAIGKIQDSLSSTASALGKLQDVVNQNAQALNTLVKQLSSNFGAISSVLNDILSRL
DKVEAEVQIDRLITGRLQSLQTYVTQQLIRAAEIRASANLAATKMSECVLGQSKRVDFCGKGYHLMSFPQ
SAPHGVVFLHVTYVPAQEKNFTTAPAICHDGKAHFPREGVFVSNGTHWFVTQRNFYEPQIITTDNTFVSG
SCDVVIGIVNNTVYDPLQPELDSFKEELDKYFKNHTSPDVDLGDISGINASVVNIQKEIDRLNEVAKNLN
ESLIDLQELGKYEQYIKWPWYIWLGFIAGLIAIIMVTIMLCCMTSCCSCLKGCCSCGSCCKFDEDDSEPV
LKGVKLHYT 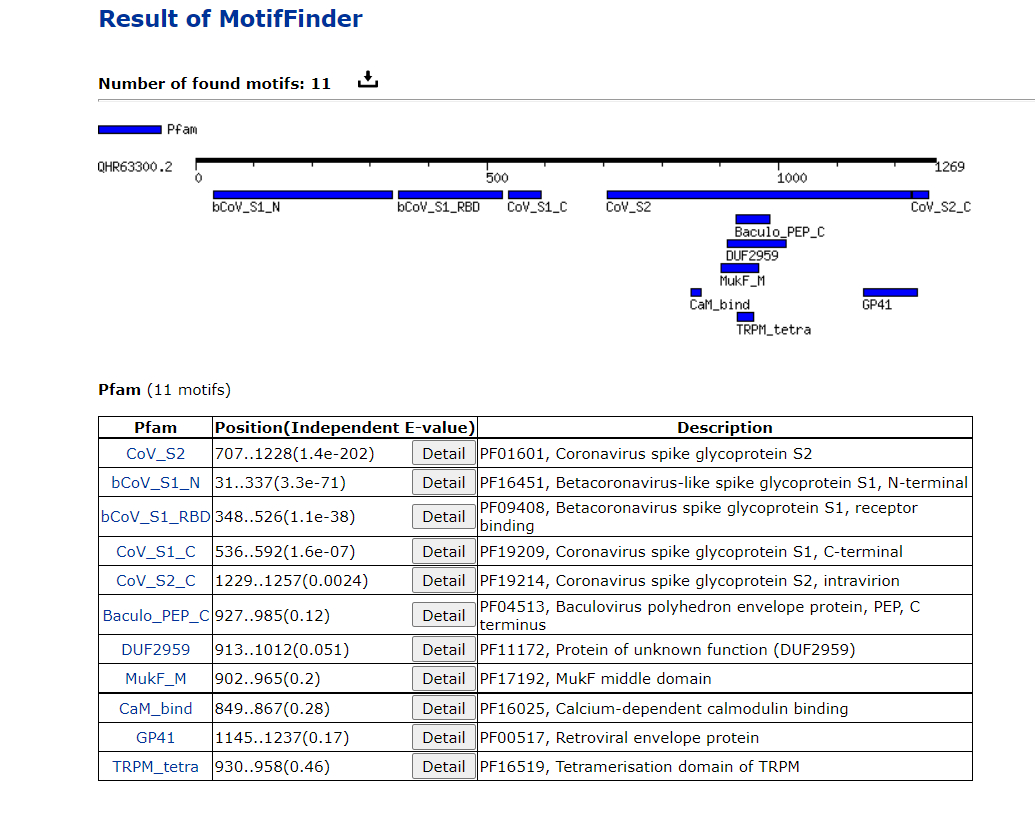
Here’s a SARS spike protein sequence and its motif pattern for comparison:
>ABD73002.1 spike glycoprotein [Severe acute respiratory syndrome-related coronavirus]
MFIFLLFLTLTSGSDLDRCTTFDDVQAPNYTQHTSSMRGVYYPDEIFRSDTLYLTQDLFLPFYSNVTGFH
TINHTFGNPVIPFKDGIYFAATEKSNVVRGWVFGSTMNNKSQSVIIINNSTNVVIRACNFELCDNPFFAV
SKPMGTQTHTMIFDNAFNCTFEYISDAFSLDVSEKSGNFKHLREFVFKNKDGFLYVYKGYQPIDVVRDLP
SGFNTLKPIFKLPLGINITNFRAILTAFSPAQDIWGTSAAAYFVGYLKPTTFMLKYDENGTITDAVDCSQ
NPLAELKCSVKSFEIDKGIYQTSNFRVVPSGDVVRFPNITNLCPFGEVFNATKFPSVYAWERKKISNCVA
DYSVLYNSTFFSTFKCYGVSATKLNDLCFSNVYADSFVVKGDDVRQIAPGQTGVIADYNYKLPDDFMGCV
LAWNTRNIDATSTGNYNYKYRYLRHGKLRPFERDISNVPFSPDGKPCTPPALNCYWPLNDYGFYTTTGIG
YQPYRVVVLSFELLNAPATVCGPKLSTDLIKNQCVNFNFNGLTGTGVLTPSSKRFQPFQQFGRDVSDFTD
SVRDPKTSEILDISPCSFGGVSVITPGTNASSEVAVLYQDVNCTDVSTAIHADQLTPAWRIYSTGNNVFQ
TQAGCLIGAEHVDTSYECDIPIGAGICASYHTVSLLRSTSQKSIVAYTMSLGADSSIAYSNNTIAIPTNF
SISITTEVMPVSMAKTSVDCNMYICGDSTECANLLLQYGSFCTQLNRALSGIAAEQDRNTREVFAQVKQM
YKTPTLKYFGGFNFSQILPDPLKPTKRSFIEDLLFNKVTLADAGFMKQYGECLGDINARDLICAQKFNGL
TVLPPLLTDDMIAAYTAALVSGTATAGWTFGAGAALQIPFAMQMAYRFNGIGVTQNVLYENQKQIANQFN
KAISQIQESLTTTSTALGKLQDVVNQNAQALNTLVKQLSSNFGAISSVLNDILSRLDKVEAEVQIDRLIT
GRLQSLQTYVTQQLIRAAEIRASANLAATKMSECVLGQSKRVDFCGKGYHLMSFPQAAPHGVVFLHVTYV
PSQERNFTTAPAICHEGKAYFPREGVFVFNGTSWFITQRNFFSPQIITTDNTFVSGNCDVVIGIINNTVY
DPLQPELDSFKEELDKYFKNHTSPDVDLGDISGINASVVNIQKEIDRLNEVAKNLNESLIDLQELGKYEQ
YIKWPWYVWLGFIAGLIVIVMVTILLCCMTSCCSCLKGACSCGSCCKFDEDDSEPVLKGVKLHYT

How Has HKU3 Been Considered Since 2020?
More recent treatments of Coronavirus information has occasionally happened upon HKU-3-3 without pinpointing anything about the unique similarities to SARS-CoV-2.
Citing Lau et al., Mahroum et al. (2022) wrote
“Investigations identified similar coronavirus in bats, SARS-related Rhinolophus bat CoV HKU3 (SARSr-Rh-BatCoV HKU3), and Chinese horseshoe bats [23]. The horseshoe bats display anti-SARS-CoV antibodies alongside the genomic sequences of SARSr-Rh-BatCoV HKU3 [24,25]. The latter with other bat coronaviruses were shown to share 88–92% nucleotide sequence homology. In fact, these studies constitute the basis for the notion that bats could potentially be the host for emerging human pathogenic coronaviruses.” From "The COVID-19 pandemic – How many times were we warned before?"
Woo et al. (2018), found that their MERS rapid detection kit could not detect HKU3, and they believed it to be SARS-like but they did indicate it as "Lineage B". The test was also negative (unable to detect) HKU1 and HKU2.
Other viruses fall closer to SARS-CoV-2 in phylogenetic analyses. This sequence of RacCS203 is from a bat in southeast Russia, and has a SARS-CoV-2-like motif pattern, but is not as similar to SARS-CoV-2 as RatG13:
>QQM18864.1 spike glycoprotein [Bat coronavirus RacCS203]
MFILLLIGYTAATTCVTGPSTENKQNVSSLMRGVYYPDDIYRSNVNVLFTVPFLRFNSTLTWYNFWNQAY
TSRVMAFGDGIYFSTVDKSNAVRGWIFGTTLDNTTQSALLINNGSLVTIQVCYFQFCANPAFLVAQNQQT
SAAVYTSSYNCTYYEVLSHIALDFTEDTGSFKRLREFVFKNYDGFLHIYGAYEPHVINIGATPSLPSTFK
PLIPLWKLSLGLNITNFKVVLTFRSNSQPLQANFAVGSLKLTTIMLSFDINGTIDKAVDCSSDPLSELKC
TLKSFNVSKGIYPTSNFRVLPSTEVVRFPNITNFCPFDKVFNATRFPNVYAWQRTKISDCIADYTVLYNS
TSFSTFKCYGVSPSKLIDLCFTSVYADTFLIRFSEVRQIAPGETGVIADYNYKLPDDFTGCVLAWNTAKQ
DIGSYFYRSHRAVKLKPFERDLSSDENGVRTLSTYDFNPNVPLDYQATRVVVLSFELLNAPATVCGPKLS
TQLVKNRCVNFNFNGLRGTGVLTDSDKRFQSFQQFGRDSADFTDSVRDPQTLQILDISPCSFGGVSVITP
GTQTSSKVAVLYQDVNCTDVPTALGLDQISAAWRVYAIGNDVFQTQAGCLVGAEHANTSHECDIPIGAGV
CASYNSPVARVGTNSIIAYEMSIGAESSIAYANNSIAIPTNFSIQVTTEVLPVSMSKTSVDCTMYICGDS
QECNKLLLQYGSFCAQLNRALSGVAVEQDKNTEAVFAQVKQIYKAPVIKDLGGFNFSQLLPDPTKPSQRS
FIEDLLFDKVTLSDAGFIKQYGDCLGDIAARDLICAQKFNGLTVLPPLLTDEMIAAYTSALVSGTATAGW
TFGIGAALQVPFAMQMAYRFNGIGVTQNVLYENQKLIANQFNSAIGKIQDSLSSTASALGKLQDVVNQNA
QALNTLVKQLSSNFGAISSVLNDILSRLDKVEAEVQIDRLITGRLQSLQTYVTQQLIRAAEIRASANLAA
TKMSECVLGQSKRVDFCGKGYHLMSFPQSAPHGVVFLHVTYVPAQEKNFTTAPAICYEGKAHFPREGVFV
SNGTHWFVTQRNFYEPQIITTDNTFVSGSCDVVIGIVNNTVYDPLQPELDSFKEELDKYFKNHTSPDVDL
GDISGINASVVNIQKEIDRLNEVAKNLNESLIDLQELGKYEQYIKWPWYIWLGFIAGLIAIIMVTIMLCC
MTSCCSCLKGCCSCGSCCKFDEDDSEPVLKGVKLHYTThis study sought to assess the level of cross-neutralization of RaTG13 afforded by antibodies raised against SARS-CoV-2 either following natural infection, vaccination or both - and found SARS-CoV-2 antibodies were neutralizing against the RaTG13 virus.
A recent study from French researchers has reported three viruses that match a bit more similar to SARS-CoV-2 than RatG13. The in bats found 530 km south of Wuhan, China, in Feuang, Laos, designated as BANAL-52, BANAL-103 and BANAL-236 (see Bat coronaviruses related to SARS-CoV-2 and infectious for human cells). Wikipedia reports higher similarity, but the study’s phylogenetic results imply closer affinity between RatG13 and SARS-CoV-2 for some of its sequence fragments. (This is important because recombination is a normal part of the transcriptional biology of betacoronaviruses).
The study authors wrote:
“Here we show that such viruses (similar to SARS-CoV-2) indeed circulate in cave bats living in the limestone karstic terrain in North Laos, within the Indochinese peninsula. We found that the RBDs of these viruses differ from that of SARS-CoV-2 by only one or two residues at the interface with ACE2, bind more efficiently to the hACE2 protein than the SARS-CoV-2 Wuhan strain isolated in early human cases, and mediate hACE2-dependent entry and replication in human cells, which is inhibited by antibodies neutralizing SARS-CoV-2. None of these bat viruses harbors a furin cleavage site in the spike. Our findings therefore indicate that bat-borne SARS-CoV-2-like viruses potentially infectious for humans circulate in Rhinolophus spp. in the Indochinese peninsula.”
Importantly, the authors also reported
“The RBDs (BANAL-237 52, -103, and -236) are closer to SARS-CoV-2 than that of any other bat strain described so far, in particular that of RaTG13, the virus identified in R. affinis from the Mojiang mineshaft where pneumonia cases with clinical characteristics a posteriori interpreted as similar to COVID-196 were recorded in 201239,40. Overall, one (H498Q (BANAL-103 and -52)) or two (K493Q and H498Q (BANAL-241)) amino acids interacting with hACE2 are substituted in these strains in comparison to SARS-CoV-2. These mutations did not destabilize the BANAL-236 / hACE2 interface, as shown by the BLI experiments (Fig. 3A) and analyzed by MD simulations.”
They concluded (incorrectly, in my opinion - evidence of recombination is only evidence of variation being moved and around and generated, NOT evidence of no selection! It’s just that their data are not inconsistent with it happening):
“Our results therefore support the hypothesis that SARS-CoV-2 could originally result from a recombination of sequences pre-existing in Rhinolophus bats living in the extensive limestone cave systems of South-East Asia and South China41,42. Many species forage in the same cave areas, including R. malayanus and R. pusillus43. In addition, the distribution of R. marshalli, R. malayanus, and R. pusillus overlaps in the Indochinese sub-region (Supp. Figure 5), which means they may share caves as roost sites and foraging habitats44. With the novel viruses here described, understanding the emergence of SARS-CoV-2 does not need to hypothesize recombination or natural selection for increased RBD affinity for hACE2 in an intermediate host like the pangolin before spillover45, nor natural selection in humans following spillover46. However, we found no furin cleavage site in any of these viruses on sequences determined from original fecal swab samples, devoid of any risk of counterselection of the furin site by amplification in Vero cells18.”
This passage although only containing the authors’ opinions, is misleading It is incorrect to say that we do not need to invoke natural selection to explain the high affinity of SARS-CoV-2 spike protein for hACE2; their data do not support this. Some affinity binding capacity is not the same as high affinity binding.
The last sentence of the passage is chilling. We already know that the WIV was routinely sampling, growing and characterizing SARS-CoV-2 like sequences. If they were growing them in Vero cells, serial passage without intent could have result in the furin cleavage site. Thus, SARS-CoV-2 laboratory origin now seems all but to have been fated, with our without direct genetic manipulation. This likelihood does not rule out direct manipulation.
For those who want to follow-up, the authors report their funding thusly:
“The work was funded by an Institut Pasteur “Covid Taskforce” and in part by the H2020 project 101003589 (RECOVER) and Labex IBEID (ANR-10-LABX62-IBEID) grants. Field and laboratory work at IP-Laos was also funded by a UK embassy grant (Grant No. INT 2021/LOV C19 02) and Luxembourg Development special grant (Grant No. LAO/030202324).”
It is worth recalling that labs around the world were involved in this research; at the time of this writing, the Pubmed database show 18 studies dealing with hACE2 and SARS virus prior to 2020.
What This All Means
While the SARS-CoV-2 novel S1/S2 cleavage motif (PPAR/S) processed by furin is uniquely associated with greatly enhanced hACE2 receptor binding affinity, the Pathogenicity Motif Pattern I reported in 2020 is associated with hACE2 receptor affinity overall. In fact, the differences between SARS and SARS-CoV-2 like viruses in terms of risk of infectivity in humans resides in this pattern.
The public’s interest in the question of laboratory or natural origin of the SARS-CoV-2 virus is immense, given the massive effect of the SARS-CoV-2 epidemic - and the massive effect of policy responses. Reasonable candidates for a potential “backbone” for SARS-CoV-2 were in fact being studied and manipulated in labs in the US and in China as far back as 2005. That’s profound because the genomes of viral lineages generated in labs are not always reported.
We need to see Dr. Baric’s full laboratory notes and correspondences with scientists in China dating back to at least 2004. We need to see any unpublished sequences over this time period.
And we need to see the same from WIV.
And labs doing research on hACE2 affinity and SARS and SARS-like sequences around the world.
That clearly is not likely to ever happen.
But at least the public now knows the degree to which gene sequence manipulations were possible dating back to 2008 and that serial passage with or without intent can lead to enhanced pathogenicity in humans; they know the was, and is, a group of so-called SARS-like sequences found in the wild indeed have some hACE2 affinity, and and this was known back in 2008; they know that the smoking gun furin cleavage site - and it is a smoking gun for origin of SARS-CoV-2 itself - is only part of the story of hACE2 receptor affinity of SARS-CoV-2 like viruses.
We would be wise to start a new moratorium on laboratory serial passage and direct genetic manipulation of potentially pathogenic viruses.
You can learn Bioinformatics @ IPAK-EDU with Dr. Lyons-Weiler in the Spring Semester and learn how to analyze RNA, DNA and protein sequences.

Thinking this may be relevant. 2005:
https://registry.opendata.aws/ncbi-covid-19/
The analysis that you did Jack, is worthy of a publication in a Virology or Genome journal. Clearly, the deniers of the lab-leak or gain-of-function research are running out of maneuvering room. The High-specificity of hACE2 to human receptors suggest a strong serial passage component in human cells. The likelihood of this kind of viral selection coming from the wild is just about nil.